

Pela primeira vez, médicos trataram com sucesso um bebê ainda no útero com um distúrbio genético raro. A criança, agora com 16 meses, está se desenvolvendo normalmente, relata um estudo de caso publicado no New England Journal of Medicine.
Ayla foi tratada com uma terapia de reposição enzimática (TRE) para doença de Pompe, que é causada pela deficiência da enzima alfa-glicosidase ácida (GAA), levando a acúmulo progressivo de glicogênio dentro dos lisossomos, com sintomas de fraqueza muscular, deterioração da função respiratória e morte prematura.
A condição tirou a vida de dois de seus irmãos, mas após a TRE no útero e o tratamento depois do nascimento, Ayla parece estar se desenvolvendo normalmente.
“O surgimento de um novo tratamento médico para aliviar o fardo da doença de Pompe para esta família e potencialmente ajudar outras famílias afetadas por doenças genéticas devastadoras é emocionante e incrivelmente satisfatório”, disse Karen Fung-Kee-Fung, especialista em medicina materno-fetal do Hospital de Ottawa (Canadá), em um comunicado. “Nós nos sentimos muito privilegiados e honrados por fazer parte desta colaboração internacional para ajudar a tornar este tratamento inédito no mundo uma realidade.”

Como funcionou o tratamento?
Quando a perda de GAA é total ou quase total, como no caso de Ayla, a condição é descrita como de início precoce. Os bebês nascidos com a doença tendem a ter corações aumentados e a maioria morrerá de complicações cardíacas ou respiratórias antes de um ano de idade se não receberem tratamento.
O tratamento padrão para a doença de Pompe é a TRE administrada a recém-nascidos por infusão intravenosa. Embora possa ajudar a diminuir o tamanho do coração, melhorar a função muscular e reduzir o acúmulo de glicogênio, não pode corrigir nenhum dano orgânico que já tenha ocorrido e muitas vezes desencadeia uma resposta imune.
Tudo isso significa que os bebês tratados para a doença de Pompe após o nascimento ainda correm risco de morte na primeira infância, ter tônus muscular ruim e dependência de ventilador. Ao fornecer TRE antes do nascimento, os médicos esperam evitar esses resultados e oferecer esperança aos bebês e suas famílias.
“A razão para dar TRE antes do nascimento é prevenir o aparecimento de danos nos órgãos, levar a enzima ao [sistema nervoso central] antes do fechamento da barreira hematoencefálica e evitar uma resposta imune à proteína ausente”, explica Tippi MacKenzie, pesquisador da UCSF (Universidade da Califórnia em San Francisco), nos EUA, e desenvolvedor do protocolo.
Após seis rodadas pré-natais de reposição enzimática no Hospital de Ottawa, Ayla nasceu. Ela está recebendo terapia enzimática pós-natal no centro de pesquisa em Ottawa. Ela tem função cardíaca e motora normais e está cumprindo os marcos do desenvolvimento infantil.
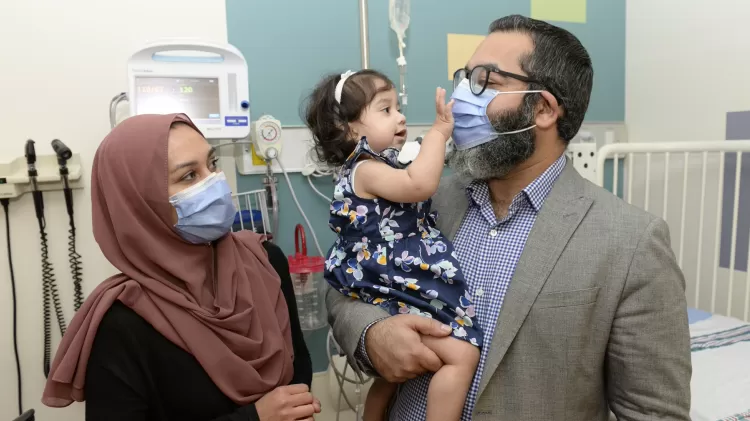
“Quando tivemos Ayla, não sabíamos se ela conseguiria andar”, disse Zahid Bashir, pai da menina. “Nós não sabíamos se ela seria capaz de falar. Não sabíamos se ela conseguiria comer. Não sabíamos se ela seria capaz de rir. Então, à medida que ela atinge cada um desses marcos, continuamos a nos surpreender com seu progresso. Então, sim, é algo que acho que às vezes podemos dar como certo, mas na maioria das vezes estamos bastante cientes de que ela é um milagre.”
Colaboração em pesquisa
O do tratamento é um feito de colaboração entre a UCSF, onde se baseia um ensaio clínico em andamento sobre o tratamento; CHEO, um hospital pediátrico e centro de pesquisa em Ottawa, o Hospital de Ottawa, onde a paciente foi diagnosticada e tratada; além da Duke University, também nos EUA, sede dos maiores especialistas do mundo em doença de Pompe.
O protocolo foi desenvolvido na UCSF. “Este tratamento expande o repertório de terapias fetais em uma nova direção”, disse o coautor sênior Tippi MacKenzie , cirurgião pediátrico do UCSF Benioff Children’s Hospitals e codiretor do Centro de Medicina de Precisão Materno-Fetal da UCSF. “À medida que novos tratamentos se tornam disponíveis para crianças com condições genéticas, estamos desenvolvendo protocolos para aplicá-los antes do nascimento.”
Em circunstâncias normais, a família da paciente teria viajado para o Centro de Tratamento Fetal do Hospital Infantil de Benioff da UCSF para participar do ensaio clínico. Quando as restrições da covid-19 inviabilizaram as viagens internacionais, especialistas dos dois hospitais canadenses e dois americanos reuniram-se com a família por vídeo para discutir alternativas.
A UCSF compartilhou o protocolo de tratamento com a equipe em Ottawa, onde a família mora. Durante todo o processo, a equipe se reuniu semanalmente por vídeo para discutir a saúde da mãe e do feto e acompanhar a resposta.
Pompe é uma das oito doenças de armazenamento lisossomal que a UCSF recebeu aprovação da FDA, órgão de controle dos EUA, para tratar com terapia de reposição enzimática no útero para um ensaio clínico de fase 1 de 10 pacientes. As outras doenças são mucopolissacaridose tipos 1, 2, 4a, 6 e 7, doença de Gaucher tipos 2 e 3 e doença de Wolman.
Os pesquisadores esperam que o sucesso desta primeira aplicação e publicação do estudo de caso aumente a conscientização sobre o ensaio clínico entre os pais com risco conhecido de transmitir essas doenças e os médicos que as tratam.
Mais dois pacientes com diferentes doenças lisossômicas foram inscritos no ensaio clínico da UCSF e ambos completaram seu curso de terapia de reposição enzimática pré-natal. A primeira paciente deu à luz no final de outubro de 2022 e a segunda no início de novembro de 2022. Ambas estão bem.
Por: Viver Bem UOL
Confira a matéria original: Clique aqui